Summary
A rapid and convenient synthesis of the psychotomimetic agent 4-iodo-2,5-dimethoxyphenylisopropylamine is described, incorporating the radioisotope 123I (T½ 13 h). With the amine function of 2,5-dimethoxyphenylisopropylamine blocked as the phthalimide, it was found that the aromatic 4-position could be directly iodinated with iodine monochloride. The phthalic acid moiety was rapidly removed with hydrazine in butanol to provide the title compound, as the hydrochloride salt, in an overall yield of 10% and in a reaction time of less than one half-life.
Introduction
4-Bromo-2,5-dimethoxyphenylisopropylamine (DOB) is known to be a centrally active agent in man1 and has been found, through labelling experiments employing 77Br and 82Br, to be actively taken up in the brain and lung of human subjects2. The relatively high energies of the gamma radiation of these two isotopes limit their usefulness as visualization agents in scintigraphic studies. Of the available isotopes of iodine, 123I emits a 159 keV gamma ray which lies within the energy range considered ideal for imaging purposes. An additional virtue is that it has a ratio of useful gamma rays to tissue dose some 50 times greater than that of 131I, an important consideration for studies in human subjects. These properties prompted the synthesis of the iodine analog of DOB, 4-iodo-2,5-dimethoxyphenylisopropylamine (DOI, 4) using 123I. However, its relatively short half-life (13 hr) requires an appropriately rapid synthetic method, the subject of this report.
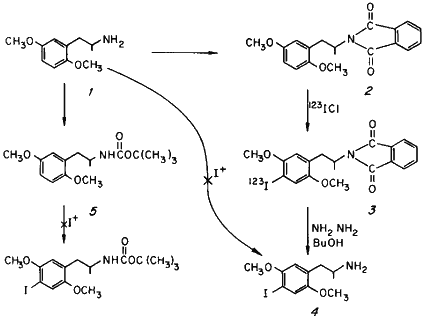
The direct iodination of 2,5-dimethoxyphenylisopropylamine 1 was tried employing a variety of procedures. The attempted direct halogenation of the amine salt of 1 with I+ or ICl, a procedure which is successful with elemental bromine1 and chlorine3 leads to preferential oxidation of the amine function. Amine derivatization as the acetamide provided protection against oxidation, but deacetylation of the iodinated intermediate could not be achieved with an acceptable yield and speed4. Protection of the amine was also possible with the easily removable tertiary butyl carbamate, but this "t-BOC" derivative could not be successfully iodinated under neutral conditions, employing the usual forms of I+. More acidic iodination conditions invariably hydrolysed the carbamate linkage of 5 preferentially, leading to decomposition.
The phthalide group was found to be sufficiently stable to allow iodination of the ring of 2 directly with iodine monochloride to provide the amide 3 which could be quickly hydrolyzed with hydrazine in butanol without isolation. The title compound 4 was then obtained by hydrolysis, as the hydrochloride salt with a radioisotopic incorporation efficiency of 10%. The identity of the incorporated halide (iodide rather than chloride) was established by chemical ionization mass spectroscopy. The in vivo distribution and brain uptake kinetics of 4 in experimental animals are reported elsewhere5,6.
Experimental
Materials and Methods
The 123ICl was obtained carrier-free from the Crocker Nuclear Laboratory of the University of California, Davis, in dilute HCl. Na131I, used for exploratory procedures, was purchased from Mallinckrodt Nuclear Corporation, St. Louis, Mo., in dilute NaOH. 1-(2,5-Dimethoxyphenyl)-2-nitropropene was purchased from the Upjohn Company. Manipulations with the isotopically labelled materials were performed in a Berkeley Box contained within a Junior Cave. Total radioactivity of the product was measured in a Squibb CRC 6a radioisotope calibrator. NMR spectra were determined on a Perkin Elmer R-32-B 60 megahertz permanent magnet spectrograph, and chemical ionization mass spectra were obtained on a AEI-MS-902 instrument. Where microanalyses are indicated only by the symbols of the elements, the results obtained were within 0.4% of the theoretical values.
2,5-dimethoxyphenylisopropylamine (1)
A solution of 38g of 1-(2,5-dimethoxyphenyl)-2-nitropropene in 200 ml THF was added to a well-stirred, refluxing suspension of 32g LiAlH4 in 750 ml THF and 75 ml anhydrous ether, at a rate that maintained reflux temperature without external heat. The addition required about 3 hr. Following 20 hr of refluxing, the suspension was cooled externally in ice, and with strong stirring there was added in sequence (against a flow of nitrogen) 32 ml H2O diluted with 2x volumes of THF, 32 ml of 15 N NaOH, and finally 96 ml H2O. Stirring was continued until the resulting suspension was completely white. The salts were removed by filtration, and the filter cake was washed with additional THF. The mother liquor and washings were pooled, and the solvent removed in vacuo to provide a residual amber oil. This crude product (36.7 g) was dissolved in 200 ml CH2Cl2, extracted with 3x150 ml dilute HCl, the pooled extracts washed with CH2Cl2, made basic with 25% NaOH, and re-extracted with CH2Cl2. The pooled extracts were washed with saturated brine, and the solvent removed in vacuo to provide a colorless oil (28.5 g). This was dissolved in anhydrous ether saturated with anhydrous HCl, filtered, washed with ether, and air-dried to provide the hydrochloride salt of 1 (27.4 g) with a yield of 70%. The mp was 115-118?C; lit. 111.5-112.5?C7; 105-106?C, increasing with hydration8.
Attempted iodination with hypoiodite ion
A solution of 1.15 g 1?HCl (5 mM) in 50 ml H2O was neutralized with NaOH and stabilized at pH 7.5 with 0.5 M phosphate buffer. With good stirring and at ambient temperature, a solution of 2.5 g KI in 50 ml pH 7.5 phosphate buffer (0.5 M) was added followed by a suspension of 4.2 g Chloramine T in 100 ml H2O containing 0.05 mol. NaH2PO4 and adjusted to a pH of 7.5 with NaOH solution using an external pH meter. The mixture became immediately opaque, developed an orange-red coloration, and within a few seconds began to deposit a dark-colored insoluble oil on the walls of the flask. After one minute a solution of 3.0 g Na2S2O4 in 50 ml H2O was added. The entire reaction mixture was made basic (pH above 10) and extracted with 3x125 ml CH2Cl2. The pooled extracts were extracted with 0.5 N H2SO4 (2x100 ml), these extracts were pooled, washed with CH2Cl2, made basic with 5% NaOH, and re-extracted with CH2Cl2. Removal of the solvent left an amber oil which was dissolved in 75 ml anhydrous ether. After standing for an hour, an insoluble gum was deposited. The solution was decanted and saturated with anhydrous HCl gas. The separated crystals proved to be unchanged 1?HCl (IR, mp and m.mp). (0.65 g, 56% recovery).
N-[1-(2,5-Dimethoxyphenyl)-2-propyl]-tert-butylcarbamate (5)
A solution of 3.90g 1 (20 mM, 3.4 ml N3CO2C(CH3)3 (20% excess), and 4.1 ml triethylamine (100% excess) In 75 ml dry THF was held at reflux on the steam-bath for 1.5h. The volatiles were removed in vacuo, and the residual oil was flooded with water (600 ml) and extracted with CH2Cl2. The pooled extracts were evaporated to a residual oil which spontaneously set to a pale yellow solid. This was recrystallized from 20 ml of boiling methanol, washed sparingly with cold methanol, and air dried to yield the product as a white crystalline solid, mp 93-94?C, 3.60 g (yield 61%).
Attempts to iodinate 5 in aqueous suspension under the hypoiodite conditions described above, or under homogenous conditions employing ICl (as described below for the phthalide derivative) resulted in no detectable nuclear iodination (as determined by the failure to incorporate 131I into the Isolated organic base fraction).
N-[1-(2,5-dimethoxyphenyl)-2-propyl]-phthalimide (2)
A suspension of 14.8 g phthalic anhydride (0.1 mol.) in 19.5g 1 as the free base (0.1 mol) was heated gradually with a soft flame to 150?C, and the temperature maintained until the evolution of water had ceased. The resulting clear amber solution was allowed to cool to about 50?C and 100 ml of hot methanol was added. The solution was stirred until homogenous, seeded with product, and cooled in an ice bath to complete crystallization. The product was removed by filtration and washed with cold methanol. Weight 24.6 g, mp 105-106?C (lit.9 mp 105.5-106?C).
4-123I-2,5-Dimethoxyphenylisopropylamine (4)
A fresh, cold solution of 0.12 ml ICl in 2.5 ml acetic acid was added directly to the aqueous 123ICl solution containing 62 mCi of 123I. The resulting pale-brown solution was added to a solution of 500 mg of 2 (1.54 mM) in 6 ml acetic acid which had been heated to 40?C with an external water bath, and stirred with a magnetic stirrer. The stirring was continued for 45 min, and then the reaction solution was quenched by pouring into 200 ml of water containing a few hundred milligrams each of KI and Na2S2O4. The colorless aqueous suspension was extracted with CH2Cl2 (3x75 ml), the extracts pooled, washed with an aqueous solution of KI and Na2S2O4, and the organic phase evaporated in vacuo. The residual colorless oil spontaneously crystallized, and was used directly in the following step without isolation or further purification. In non-radioactive runs, this product, N-[1-(4-iodo-2,5-dimethoxyphenyl)-2-propyl]-phthalimide 3 was isolated and characterized. Fine white crystals from methanol, mp 103-103.5?C. Mixed melting point with 2, 85-98?C.
The intermediate compound 3 was dissolved in 10 ml n-butanol containing 0.5 ml 95% NH2NH2. The clear solution was placed in a boiling water bath and swirled with occasional venting as needed. The solution became progressively cloudy and developed a yellow-brown color. After a few minutes, the cloudiness cleared, there was a deposition of a cottage cheese-like precipitate, and a concurrent fading of the color. An additional 2 ml of butanol was added, and the heating continued for a total of 15 min. The reaction mixture was cooled and the solids broken up under dilute HCl. The resulting suspension was filtered, and the solids washed with additional HCl. The combined mother liquor and washings were washed with methylene chloride (discarded), made basic with NaOH (pH above 9), and extracted with CH2Cl2 (3x50 ml). The combined extracts were washed with water, decanted to a fresh dry centrifuge tube to remove particulate water, and extracted with 3.5 ml 0.1 N HCl. The phases were separated by centrifugation, and the aqueous phase removed for sterilization (by millipore filtration) and radioactivity assay. The retained activity in the final product was 3.6 mCi representing a specific activity of 35.7 mCi/mM available at the time of animal administration. Non-radioactive preparations provided samples of 4 which were characterized physically and chromatographically. The properties of these specimens were identical in all respects with those reported9.