Abstract
The syntheses of 122I- and 125I-labelled 2,4-dimethoxy-N,N-dimethyl-5-iodophenyl- isopropylamine, 3,5-dimethoxy-N,N-dimethyl-2-iodophenylisopropylamine and 2,6-dimethoxy- N,N-dimethyl-3-iodophenylisopropylamine are described. The speed (3 min, including purification) and yield (45-85%) obtained in direct iodination procedures utilizing chloramine-T have allowed the use of the short-lived positron emitter 122I (t½ = 3.6 min) in brain blood flow imaging studies. The three appropriate precursors (the meta-dimethoxy-N,N-dimethylphenylisopropylamines) were prepared from the corresponding phenylacetone analogues by reductive amination employing dimethylamine and NaCNBH3. The ketones were obtained from the appropriate nitrostyrenes through reduction with elemental Fe.
Introduction
Two classes of imaging agents for brain blood flow determination exist. One has the properties of high extraction by brain tissue on the first pass and retention by brain for a time period sufficient for imaging; the other depends upon free diffusion in and out of brain tissue. The first example of a retained agent was 4-82Br-2,5-dimethoxyphenylisopropylamine (4-82Br-2,5-dimethoxyamphetamine)1. Two similar iodinated analogues have been described2,3, and a chemically related diamine has also been utilized for brain imaging studies4. These compounds were prepared with gamma-emitting isotopes which were inappropriate for positron emission tomography (PET). [...]
Previously, considerable synthetic time has been required for the incorporation of the radio-iodine into the iodinated amphetamine analogues, either because derivatization was needed to protect the amine from oxidation2 or a slow halogen-halogen exchange process was used3,4. Syntheses utilizing 123I (t½ 13 h) or 131I (t½ 8 d) can be performed with little loss of the radionuclide in these relatively slow processes; iodinations involving 122I require fast synthetic routes. Iodination of the 2,5-dimethoxy-N,N-dimethylamphetamine with ICl required high temperature, an organic reaction medium and preformation of radio-labelled ICl7. The para-dimethoxy orientation of this 2,5-dimethoxy analogue did not activate the ring sufficiently for direct electrophilic iodination by methods such as Chloramine-T8. The syntheses of the corresponding meta-dimethoxy-N,N-dimethyl-amphetamine counterparts are described here. These compounds have proven sufficiently activated to allow direct radio-iodination employing Chloramine-T in an aqueous medium.
Scheme 1
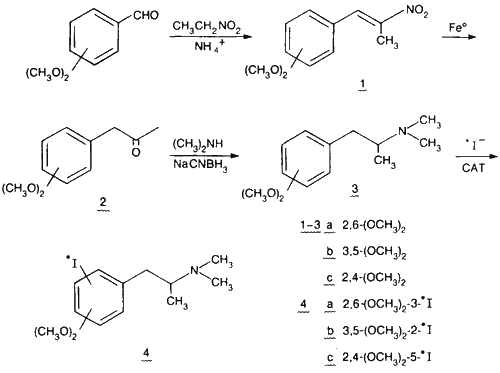
The synthetic procedures leading to the three meta-substituted compounds were the same, starting with appropriate precursors, and are outlined in Scheme 1. Dimethoxybenzaldehyde was reacted with nitroethane (as both reagent and solvent) with a catalytic amount of ammonium acetate via the Knoevenagel reaction9. The resulting nitrostyrene 1 was reduced to the phenylacetone 2 with elemental iron in acetic acid10. Reductive amination of the ketone with dimethylamine and sodium cyanoborohydride11 yielded the tertiary amine 3 which was iodinated directly in an aqueous system containing either 122I-iodide or 125I-iodide and Chloramine-T (CAT).
The resulting 122I-labelled meta-dimethoxy-N,N-dimethyl-iodoamphetamines 4 have been utilized in PET studies of cerebral perfusion in mongrel dogs. These agents demonstrate rapid brain uptake and long term retention in cerebral tissue and show promise as brain blood flow radiopharmaceuticals12,13.
Experimental
2,6-Dimethoxybenzaldehyde was prepared by the procedure of Lambooy14 except that butyllithium was used to form the lithiation product with 1,3-dimethoxybenzene (Aldrich Chemical Co.) followed by reaction with N-methylformanilide. [...] Distillations were performed using a Kugelrohr apparatus at the temperatures and vacuum pressures indicated.
2,6-Dimethoxy-β-methyl-β-nitrostyrene. 1a.
To a solution of 10.0 g of 2,6-dimethoxybenzaldehyde in 50 ml nitroethane there was added 0.5 g anhydrous ammonium acetate, and the mixture was held on the steam bath for 2 h. The solvent was removed in vacuo giving a heavy reddish oil which, upon dissolving in 25 ml hot methanol and cooling, yielded bright yellow crystals, 12.0 g (90% yield), mp 101.5-102.5?C.
In the same manner, 3,5-dimethoxy-β-methyl-β-nitrostyrene (1b) was prepared (94%), mp 87-88?C (lit. mp 88?C15), as well as 2,4-dimethoxy-β-methyl-β-nitrostyrene (1c, 77%), mp 78-79?C (lit. mp 76-78?C16).
2,6-Dimethoxybenzylmethyl ketone. 2a.
A solution of 11.5 g of 1a in 80 ml warm acetic acid was added to a suspension of 35 g of electrolytic iron dust in 150 ml acetic acid. The mixture was heated on the steam bath until a vigorous reaction set in. The resulting paste was diluted with another 40 ml acetic acid and heated for an hour. The reaction was quenched in 1.5 L water with stirring, decanted from unreacted Fe and extracted with 3x100 ml methylene chloride. The pooled extracts were washed with 50 ml 5% NaOH and the solvent removed in vacuo to yield 10.5 g of a pale amber oil. This was distilled in vacuo giving 8.7 g (86% yield) of a colorless oil (95-105?C/0.4 mmHg).
In the same manner, 3,5-dimethoxybenzylmethyl ketone (2b, bp 110-130?C/0.3 mmHg, 83%), and 2,4-dimethoxybenzylmethyl ketone (2c, bp 125-145?C/0.5 mmHg, 65%) were prepared, both as colorless oils.
2,6-Dimethoxy-N,N-dimethylphenylisopropylamine. 3a.
A solution of 7.6 g of 2a in 100 ml methanol was added to a warm solution of 25 g dimethylamine hydrochloride in 60 ml methanol. With vigorous stirring there was added 3.3 g NaCNBH3, followed by conc. HCl dropwise as needed to maintain the reaction medium at a pH of about 6. When acid was no longer required (about 48 h) the methanol was removed in vacuo and the residue poured into 2 L of dilute sulfuric acid. The mixture was extracted with 2x100 ml methylene chloride (discarded), made basic with 25% NaOH and reextracted (3x100 ml methylene chloride). The pooled extracts were stripped of solvent, giving 2.38 g of a colorless oil which was distilled (110-120?C/0.4 mmHg), yielding 1.49 g (17% yield) of a white oil. The perchlorate salt was recrystallized from isopropanol and ether, m.p. 109-110?C.
In the same manner, 3,5-dimethoxy-N,N-dimethylphenylisopropylamine (3b) was prepared, 3.3 g, from 7.4 g of the ketone (39% yield), m.p. of the perchlorate salt 100-101?C.
In the same manner, 2,4-dimethoxy-N,N-dimethylphenylisopropylamine (3c) was prepared, 10.6 g from 12.4 g of the ketone (74% yield), m.p. of the perchlorate salt 98-98.5?C.
2,4-Dimethoxy-N,N-dimethyl-5-125I-phenylisopropylamine. 125I-4c.
[...] (µg synthesis, see the article's [full text] for details)
Employing the above optimum conditions, 2,6-dimethoxy-N,N-dimethyl-3-125I-phenylisopropylamine (125I-4a) was prepared (50% yield in 1 min).
In the same manner, 3,5-dimethoxy-N,N-dimethyl-2-125I-phenylisopropylamine (125I-4b) was prepared (83% yield in 1 min).
2,6-Dimethoxy-N,N-dimethyl-3-122I-phenylisopropylamine. 122I-4a.
[...] (µg synthesis, see the article's [full text] for details)
The assignment of the iodine substitution position and the chromatographic characteristics of nonradioactive 4a were established by a separate synthesis employing millimolar quantities of sodium iodide and 3a. To a solution containing 150 mg (0.67 mmole) 3a and 120 mg (0.80 mmole) NaI in 30 ml 0.25 M H3PO4 there was added 229 mg (1.0 mmole) CAT. The reaction was allowed to proceed at 60?C for 5 min and was quenched with 300 mg (1.6 mmole) Na2S2O5. The solution was made basic with NaOH and extracted with CH2Cl2 (3x30 ml). The iodinated product (46% yield) was separated from the starting material (3a) and side products by semi-preparative HPLC. The HPLC and TLC chromatographic . characteristics of 4a were identical to 125I-4a and 122I-4a. The major side-product (20% yield) using a stoichiometric excess of CAT in these nonradioactive procedures proved to be the 3-chloro analog (3-chloro-2,6-dimethoxy-N,N-dimethylphenylisopropylamine) as established by NMR of a chromatographically separated sample. The positional assignment of both halides (as shown by NMR, q.v.) is the same as that reported for the bromination of 2,6-dimethoxyphenylisopropylamine17.
In the same manner, 3,5-dimethoxy-N,N-dimethyl-2-122I-phenylisopropylamine (122I-4b) was prepared (68% incorporation of 122I removed from the loop). The synthesis of nonradioactive 4b (60% yield) was conducted as with 4a. The chromatographic characteristics of 4b were identical to 125I-4b and 122I-4b.
In the same manner, 2,4-dimethoxy-N,N-dimethyl-5-122I-phenylisopropylamine (122I-4c) was prepared (85% incorporation of 122I removed from the loop). The synthesis of nonradioactive 4c (65% yield) was conducted as with 4a. The chromatographic characteristics of 4c were identical to 125I-4c and 122I-4c.
A summary of the yields of 125I- and 122I-labelled 4a, 4b and 4c and the chromatographic data is given in Table 3.