Introduction
Mescaline [I] is a simple base occurring in various cacti of the genus Anhalonium and has been prepared synthetically by a number of methods.1 Its pharmacological actions were first described by Lewin2 in 1894 and are characterised chiefly by stimulation, euphoria, and visual hallucinations, which last take the form of fantastically beautiful and constantly changing patterns of every shade and colour. These properties are, no doubt, the basis of the widespread use of dried slices of the cacti (mescal buttons) in the religious rites of native tribes in Mexico and Central America where the plants are indigenous. An excellent account of the ritual use of the drug is given in the monograph by Rouhier.3 In 1940, Tayleur Stockings4 pointed out the similarities between the mental phenomena induced by mescaline and those in certain types of naturally occurring psychosis and stressed the importance of the study of drugs having actions similar to mescaline as possible aids to the better understanding of the nature of mental disorders.
Mescaline is a true central nervous stimulant, a property uncommon in simple phenylethylamine derivatives. It is well known, however, that conversion of the ethylamine side chain to isopropylamine by substitution of a methyl group on the α (to -NH2) carbon atom leads to the production of a group of compounds many of which have, in varying degrees, central nervous stimulant activity. It therefore appeared to be a matter of interest to determine the effect of a similar substitution in mescaline itself and the synthesis of 1-(3,4,5-trimethoxyphenyl)-isopropylamine [II] was undertaken. The present paper gives an account of the methods employed for this synthesis. The pharmacological properties of this new homologue are being investigated and will be described at a later date.
Routes For Synthesis
Of several possible routes for the synthesis two have been examined and are depicted schematically below.
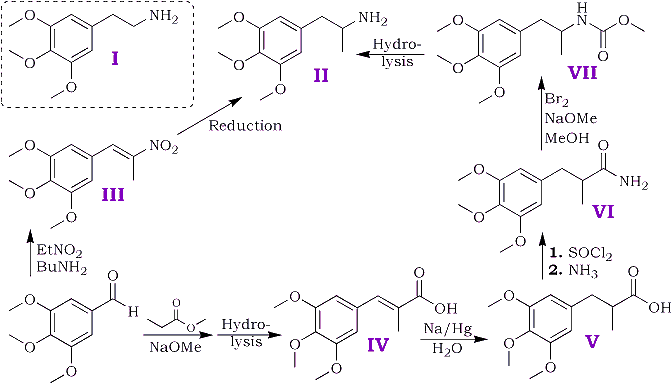
3,4,5-trimethoxybenzaldehyde was prepared from 3,4,5-trimethoxybenzanilide by the method of Sonn and Müller5 with the modifications introduced by Cook et al.6; consistent yields of 70-75% were obtained in all runs. Other methods were tried but were found unsatisfactory. Direct reduction of 3,4,5-trimethoxybenzoyl chloride with gaseous hydrogen and a "poisoned" palladium catalyst by Rosenmund's7 method led to the production of a mixture of 3,4,5-trimethoxybenzyl alcohol and 3,4,5-trimethoxytoluene. The preparation from 3,4,5-trimethoxybenzonitrile by Stephen's8 method gave negligible yields of aldehyde (cf. Baker and Robinson9).
The above aldehyde condensed readily with nitroethane giving an almost quantitative yield of 1-(3,4,5-trimethoxyphenyl)-2-nitropropene [III]; n-butylamine was found preferable to piperidine as the condensing agent.
Attempts to reduce the nitrostyrene [III] by catalytic methods using a palladium-charcoal catalyst and a mixture of methyl alcohol and acetic acid as solvent resulted in a rapid uptake of six of the eight hydrogen atoms required by theory, after which all reaction ceased. Reduction by zinc dust and acetic acid followed by sodium amalgam as described by Späth1a in his synthesis of mescaline gave a poor yield (10%) of the required amine. Electrolytic reduction at a lead cathode as described by Slotta and Szyszka1c in an analogous synthesis of mescaline gave a satisfactory yield.
Owing to the initial difficulties in the reduction of the nitrostyrene the alternative route was examined. The overall yield (calculated on 3,4,5-trimethoxybenzaldehyde) was less by this route owing to the failure to find satisfactory conditions for the conversion of the amide [VI] to the amine.
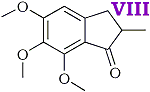
2-methyl-3-(3,4,5-trimethoxyphenyl)-propenoic acid [IV] was obtained in good yield by condensing 3,4,5 trimethoxybenzaldehyde with methyl propionate and hydrolysing the ester produced in the condensation. The unsaturated acid was reduced quantitatively to 2-methyl-3-(3,4,5-trimethoxyphenyl)-propanoic acid [V] by treatment, in aqueous solution, with sodium amalgam. Treatment of the saturated acid with thionyl chloride gave the corresponding acid chloride but attempts to purify the latter by distillation appeared to lead to ring closure to 2-methyl- 5,6,7-trimethoxyhydrindene-1-one [VIII] bp 195-200?C/0.6 mmHg. Crystallisation from acetone gave colourless plates, mp 86-87?C. The 2,4-dinitrophenylhydrazone crystallised from acetic acid in small garnet-coloured crystals, mp 180?C.
Treatment of the crude acid chloride with concentrated aqueous ammonia or finely powdered ammonium carbonate led to extensive hydrolysis and little amide production. Reaction with dry gaseous ammonia in ethereal solution gave a satisfactory yield of the required amide [VI].
The ordinary Hoffman procedure for degradation of the amide to the required amine gave only traces of basic products and an alternative route described by Jeffreys10 for certain aliphatic amines was tried. In this method the isocyanate formed by the Hoffmann method reacts with methyl alcohol to give a urethane (in this case [VII]) which is then hydrolysed to the amine.
The urethane proved somewhat resistant to hydrolysis and the yield of amine obtained was small. Since the electrolytic method for the reduction of the nitrostyrene in the first route proved both expeditious and satisfactory the second route has not been the subject of further experiment.
Experimental
1-(3,4,5-trimethoxyphenyl)-2-nitropropene
3,4,5-trimethoxybenzaldehyde (39.2 g) and nitroethane (15.7 g) were dissolved in ethyl alcohol (30mL) by gentle warming (40?C); 1.5 ml. of n-butylamine were added and the container closed and kept at 35-40?C. for seven days. The condensation product formed a supersaturated solution but crystallisation was easily induced by cooling and scratching the sides of the vessel when long yellow needles separated out and were collected by filtration. The product was almost pure. Yield 48 g. (95% of theory). A small additional yield of less pure material was obtained by cautious evaporation of the mother liquor. Recrystallisation from ethyl alcohol gave stout, bright yellow prisms, mp 95?C.
dl-1-(3,4,5-trimethoxyphenyl)-2-aminopropane hydrochloride
The apparatus used for the electrolytic reduction of the nitrostyrene was similar to that described by Slotta & Szyszka1c. A 500-ml. beaker was used as the cell and was lined with pure sheet lead 20x10 cm forming the cathode. The anode, also of sheet lead, was enclosed in a porous cell 5x15 cm standing in the centre of the beaker. Cooling was provided by a thin walled gas washing bottle standing in the porous cell and an external bath, water being circulated through both. The anode compartment was filled with dilute (10%) sulphuric acid and the cathode compartment with a mixture of acetic acid (100 ml), ethyl alcohol (100 ml) and hydrochloric acid (50 ml) and a small stirrer and thermometer placed in position in it. The finely powdered nitrostyrene (25.3 g) was placed in the cathode compartment and the stirrer started. A current of 5-6 amperes (7-8 volts required), equivalent to 25-30 mA/cm3 of cathode surface, was passed; if the temperature rose above 40?C the current was interrupted until the cell had cooled. Reduction took place steadily, little hydrogen being evolved, while the nitrostyrene went into solution and the latter lost its yellow colour. When the hydrogen liberated at the cathode was about 25%, in excess of the amount required by theory for the reduction, the rate of evolution increased rapidly. A small amount of a pale yellow amorphous substance remained undissolved. The solution in the cathode compartment was removed and concentrated under reduced pressure and the residue diluted with water and filtered. The filtrate was extracted several times with chloroform to remove non basic products. Basification of the residual aqueous solution, with sodium hydroxide, precipitated the base as an oily liquid which was then extracted with chloroform. Evaporation of the chloroform solution left an oily liquid with a faint "basic" odour very like that of mescaline itself; it absorbed carbon dioxide rapidly from the air, forming a solid carbonate. Treatment of a concentrated chloroform solution of base followed by the removal of the solvents left a white residue of the hydrochloride which was recrystallised from methanol forming small leaflets, mp 219-220?C. The mother liquors were evaporated and the residue recrystallised again. Total yield 12.2g (47% of theory).
2-Methyl-3-(3,4,5-trimethoxyphenyl)-propenoic acid
Dry, finely powdered, sodium methoxide (9 g) was suspended in dry methyl propionate (56 g) cooled below 5?C. by an ice bath. Finely powdered 3,4,5-trimethoxybenzaldehyde (19.6 g) was added in small portions, with mechanical stirring, as rapidly as it dissolved (30 min.). Stirring was discontinued and the mixture set to a soft paste which was allowed to come to room temperature overnight and then heated at 60-70?C. for two hours on the following day. Water (100 ml) and acetic acid (12 ml) were added and the stirrer run slowly until the pasty mass had broken up and the mixture separated into two layers. The upper (ester) layer was separated off and as much as possible of the excess methyl propionate removed by distillation. The residue was hydrolysed by refluxing with a slight excess of N/1 methyl alcoholic potassium hydroxide. The methyl alcohol was removed by distillation and the residual crude potassium salt boiled with water (charcoal) and filtered; acidification of the filtrate precipitated the required acid which was collected and recrystallised from diluted acetone, forming long, strongly flattened, iridescent plates, mp 156.5?C. Yield (recrystallised material) 20.6 g (82% of theory).
dl-2-methyl-3-(3,4,5-trimethoxyphenyl)-propanoic acid
2-methyl-3-(3,4,5-trimethoxyphenyl)-propenoic acid (25.2g) was dissolved in N/2 sodium hydroxide (200 ml.) and the solution stirred gently while sodium amalgam (4%, 200 g.) was added over a period of two hours. When all the amalgam had reacted the aqueous layer was separated and filtered. Cautious acidification of the filtrate precipitated the required acid which was collected and recrystallised from benzene forming small colourless rhombic plates, mp 116?C. Rapid acidification of the filtrate led to the acid separating as a hard granular mass which was much more difficult to purify, due to mother liquid trapped in it.) Yield (recrystallised material) 25 g (98.5% of theory).
dl-2-methyl-3-(3,4,5-trimethoxyphenyl)-propionamide
The above saturated acid (12.7 g) and thionyl chloride (6.0 g) were heated together under a reflux condenser for thirty minutes and the crude acid chloride dissolved in dry chloroform (50 ml). Dry ether (200 ml) was cooled to 0?C. and stirred vigorously while a rapid stream of dry ammonia was passed into it. The chloroform solution of the acid chloride was added slowly while the stream of ammonia was maintained. When all the acid chloride had been added, the solvents were distilled off on the water-bath and the residue extracted with cold water to remove the ammonium chloride. The crude amide was recrystallised (charcoal) from methyl alcohol (60%) forming small prisms; mp 141?C. Yield (recrystallised material) 11.4 g (90% of theory).
dl-1-(3,4,5-trimethoxyphenyl-2-aminopropane hydrochloride (second method)
The above amide (5.06g) was dissolved in dry methyl alcohol (100 mL) and sodium (46 g) dissolved in dry methyl alcohol (25 ml.) was added and the mixture cooled to 0?C. Bromine (3.2 g), diluted with twice its volume of dry methyl alcohol, was added all at once, with rapid stirring. A further amount of sodium 0.46g) was then added in the same way as the first and the reaction mixture heated at 60?C for two hours. The methyl alcohol was partly removed by distillation and the residue diluted with water. A brown oil separated out and slowly solidified at room temperature and which was presumably the crude urethane (VII). The solid was filtered off and boiled for eight hours with 20 per cent. (constant boiling) hydrochloric acid. After cooling the reaction mixture was extracted several times with chloroform to remove non basic impurities. Basification of the residual aqueous liquid precipitated the base which was extracted and converted to the hydrochloride as in the previous preparation; mp and mixed mp with the material from reduction of the nitrostyrene, 219?C. Yield 0.81g (15% of theory).
Summary
- A satisfactory method for the synthesis of dl-1-(3,4,5-trimethoxyphenyl)-2-aminopropane, a compound closely related to mescaline, is described.
- In the course of the synthesis the following additional new compounds were prepared:
- 1-(3,4,5-trimethoxyphenyl)-2-nitropropene
- 2-methyl-3-(3,4,5-trimethoxyphenyl)-propenoic acid
- 2-methyl-3-(3,4,5-trimethoxyphenyl)-propanoic acid
- 2-methyl-3-(3,4,5-trimethoxyphenyl)-propionamide
- It is probable that a sixth new compound, viz. 2-methyl-5,6,7-trimethoxyhydrindene-1-one was formed at one stage of the synthesis by ring closure of 2-methyl-3-(3,4,5-trimethoxyphenyl)-propionyl chloride.
It is well known that alkoxy groups greatly facilitate ring closures of this type. The material forms a 2,4-dinitrophenylhydrazone and analytical figures for this and the original compound are in reasonable agreement with the above assumption.